How did We work for the Discovery of Candidate Drug Molecules?
As I mentioned before, this project aims to develop a drug against viral ds-DNA, which is associated with Herpes Simplex Virus, Cytomegalovirus, Adenovirus type 5, SV40 Polyoma virus and Vaccinia virus.If viral DNA is blocked, it cannot replicate inside the cell. Targeting virus DNA is more accurate, but it can also bring disadvantages.
In our study
We use the well-known TAT from HIV ; I24 is a 9-mer amino acid sequence that can be fused to TAT. Here, random mutations were obtained by making single-point mutations in the sequence of TAT-I24. New mutant peptide sequences obtained by single point mutations were subjected to MD simulations (250 ns).
After the MD simulations, the MM-GBSA and MM-PBSA binding free energies were calculated. Molecules with high MM-PBSA and MM-GBSA scores can be used as drug candidates in the next step.
While doing MD simulations
Molecular Dynamics Simulation (MD) tests the interactions of molecules in the system for a certain period of time by making calculations.In the MD simulation performed here, DNA and peptides were tested in a explicit water solvent and simulated for 250 ns. Therefore, the interactions of atoms and molecules during this time were tested. We benefited from AMBER/20 and GPU performance while doing MD simulation. In this way, we performed more MD simulations in a shorter time. As a result of MD simulations, 250 frames were obtained. We viewed them in a video. We observed that structural changes occurred between molecules with good MM-PBSA and MM-GBSA (ΔG) scores. There were particularly strong interactions between ds-DNA and mutant ligands.
MM-GBSA and MM-PBSA Calculations
After MD simulations, MM-GBSA and MM-PBSA binding free energies are calculated. Here; MM-PBSA uses the Poisson-Boltzmann equation to calculate the electrostatic contribution to free energy; MM-GBSA uses the Generalized Born approach. However, we cannot compare the speed between MM-GBSA and MM-PBSA. This may vary depending on the parameters we use. The lower the energy obtained in these Computational experiments, the stronger the interaction between the molecules. Therefore, TAT-M178 gave the best results with -103 kcal/mol (MM-GBSA) and -76.6 kcal/mol (MM-PBSA).
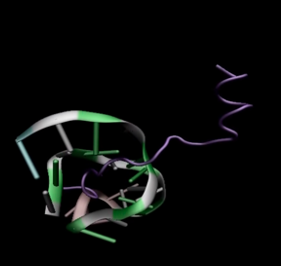
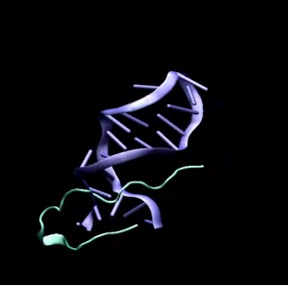
We had 120 variants of TAT-I24. We performed MD simulation for each of this variants. As a result, TAT-M178 gave the best score.
In Figure 2 (Frame:0) , that is, they are not fully interacting at the MD starting point, but in Figure 3(Frame:100) , There is already strong interaction between TAT-M178.
I am doing my master's degree in Tissue Engineering and Regenerative Medicine at Bahçeşehir University / Faculty of Medicine. I also work in the Computational Biology and Molecular Simulation Laboratory (Durdagi Research Group (durdagilab.com)). Project 1: Software Based Drug Development Project: 'Discovery of Paxlovid Analogs as SARS-CoV-2 Main Protease Inhibitors'. Project 2: Structure of human Bruton's Tyrosine Kinase-associated protein virtual screening and reconstruction of target drug molecules, (Covdock-MD-MMGBSA at Schrödinger) Project 3: HPC-Derived Affinity Enhancement of Antiviral Drugs

Great work Ezgi!
thank you 🙂 Im really happy