Improving existing genomic tools for HPC infrastructure

Project reference: 1817
Since the emergence of the Next-Generation Sequencing technologies the number of genomics projects has risen exponentially. Human population genomics is an exciting field for research with significant outputs into clinical medicine. While there was just one human genomic sequence in 2001, we now have more or less complete and de novo assembled genomes from about 50 individuals. Eventually, the overall population variation is well sampled thanks to “1000 Genomes Project”, “100,000 Genomes Project (Genomics England)” and “100,000 Genomes (Longevity)”. Although many tools exist to perform sub-optimal short read sequence alignment, variant calling and phasing, there is still a lot about them to be improved. For example, the existing tools often run in a single-threaded mode and only on the main CPU and often need hundreds of GB of RAM.
The suggested project will improve existing software tools widely used in genomics, especially population genomics.
The main focus will be to contribute to the development of bcftools package (http://samtools.github.io/bcftools/howtos/index.html) in any of the following (ordered with increasing difficulty):
- Improve automated tests, fix existing bugs, improve documentation;
- Improve efficiency of the implementation;
- Improve the built-in query language;
- Collect examples of incorrect alignment from publicly available datasets (for future realignment algorithm and implementation);
- Conceptually implement IndelRealigner (is to be dropped as a standalone tool from GATK bundle since version 4.0) into “samtools mpileup” or a separate package (so there won’t be a standalone tool anymore which could be used by other tools for comparisons/benchmarking; parts of IndelRealigner will remain in the HaplotypeCaller,however, it is not possible to call the routines from external (3rd-party tools));
- Develop a tool to compare VCF/BCF files (a plain UNIX diff does not work practically and SQL-based approaches are too slow). Complex mutations currently need to be split into individual/atomic changes prior to any comparison (“bcftools norm”). An ideal tool would even respect haplotypes and multi-sample VCF/BCF files.
Alternatively, the project will focus on improving java-based GATK (https://software.broadinstitute.org/gatk), IGV (https://github.com/igvteam/igv) or Picard (http://broadinstitute.github.io/picard) tools, or FreeBayes (C++, https://github.com/ekg/freebayes), depending on the preferences of the applicant. Interested candidates should look through currently opened Github Issues, browse the sources and read briefly through the documentation.
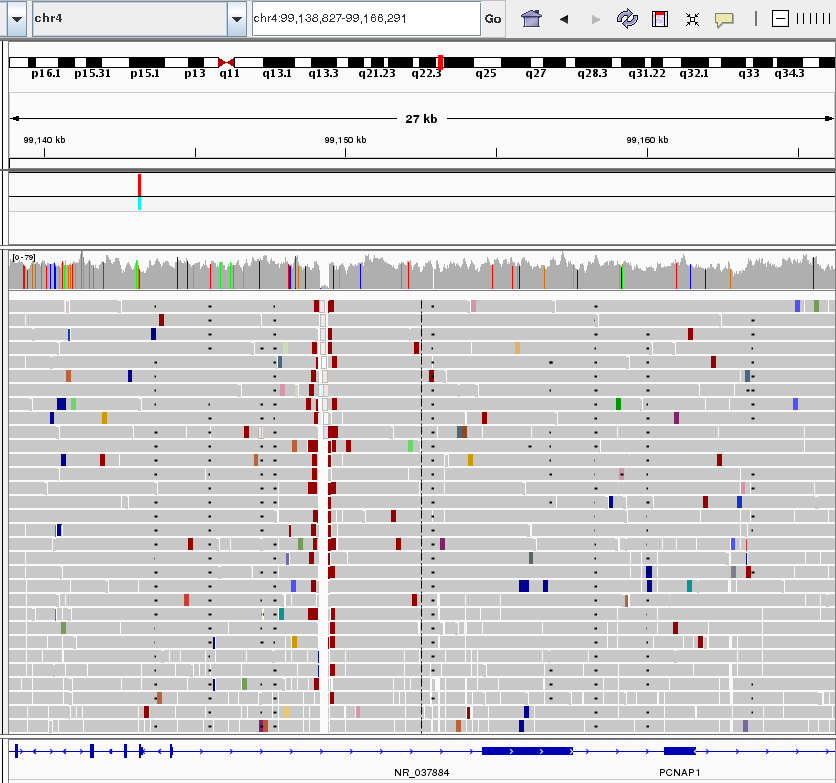
A view of short sequence reads aligned to the human genome reference sequence rendered by IGV software. A low-covered region with a deemed deletion in the chromosome of a proband is visible as a vertical gap. Reads are coloured according to their insert size and orientation. Short black lines (appearing almost as dots) in several columns represent gaps in the sequence alignment of the individual raw sequencing reads.
Project Mentor: Martin Mokrejš, Ph.D.
Project Co-mentor: Petr Daněček, Ph.D.
Site Co-ordinator: Karina Pesatova
Learning Outcomes:
The student will learn about bioinformatic tools, algorithms, genomics, improve his/her programming skills.
Student Prerequisites (compulsory):
MSc. in bioinformatics or computer sciences, proficient in either of C, C++, java, advanced git knowledge, good software programming practice, automated test-suites, Github, TravisCI. Working knowledge of basic genomic concepts. Candidates should become reasonably familiar with the topic, the tools to be worked on and training materials before interview.
Student Prerequisites (desirable):
Statistical genetics, software performance profiling/debugging.
Training Materials:
Documentation, Tutorials, Examples, Testing/Demo data available on the internet.
- http://samtools.github.io/bcftools
- https://software.broadinstitute.org/gatk
- https://github.com/igvteam/igv
- https://github.com/ekg/freebayes
- http://seqanswers.com
- https://www.biostars.org
- https://en.wikipedia.org/wiki/Sequence_alignment
- https://en.wikipedia.org/wiki/Smith–Waterman_algorithm
Workplan:
- 1st week: Training week.
- 2nd week: Read documentation, code and test examples, test the tools, fix known but simple bugs (e.g. github pull requests).
- 3rd week: Sum up the work plan.
- 4-7th week: Do the main development work.
- 8th week: Finish the development, document the work, that’s been done write a Final report.
Final Product Description:
The output will be in the form of software documentation, automated tests, accepted patches to existing code fixing bugs or implementing new features.
Adapting the Project: Increasing the Difficulty:
Example topics (see Abstract) are ordered with increasing difficulty. Similar listing will be made if work on another tool(s) is taken. The student will work on more difficult tasks as the time will permit or work on simpler tasks related to another software tool.
Adapting the Project: Decreasing the Difficulty
The student will start with simpler tasks (an example is listed in the beginning of the project description) and continue with more difficult tasks, if possible. Alternatively, will work on simpler tasks on another software tool.
Resources:
The work will utilize Linux-based workstations, remote work on two RedHat-based supercomputer clusters with “PBS Pro” scheduler. Ability to work with bash, git and proficiency in any of the C, C++, java is required for successful work on the project.
Organisation:
IT4Innovations national supercomputing center
![]()
