Density Functional Theory – What is it?

How are the materials made? How do electrons and ions arrange in a stable structure? Is your material a metal or an insulator? Is it opaque or transparent? Finally, is it a good nuclear fuel?
Density Functional Theory (DFT) is a computational quantum mechanical modelling method used to solve the microscopic problem starting from the charge density of our system. Electrons and Ions in a solid experience attractive interactions, electron-ion (negative-positive charges), and repulsive ones, electron-electron (negative-negative charges). Considering still Ions, interactions and focusing on charge density, it is possible to solve the electronic problem with DFT: given the crystal structure and number of valence electrons in a few second you will obtain the equilibrium distance and all the electrons available states, which ultimately determine the main properties of the material.
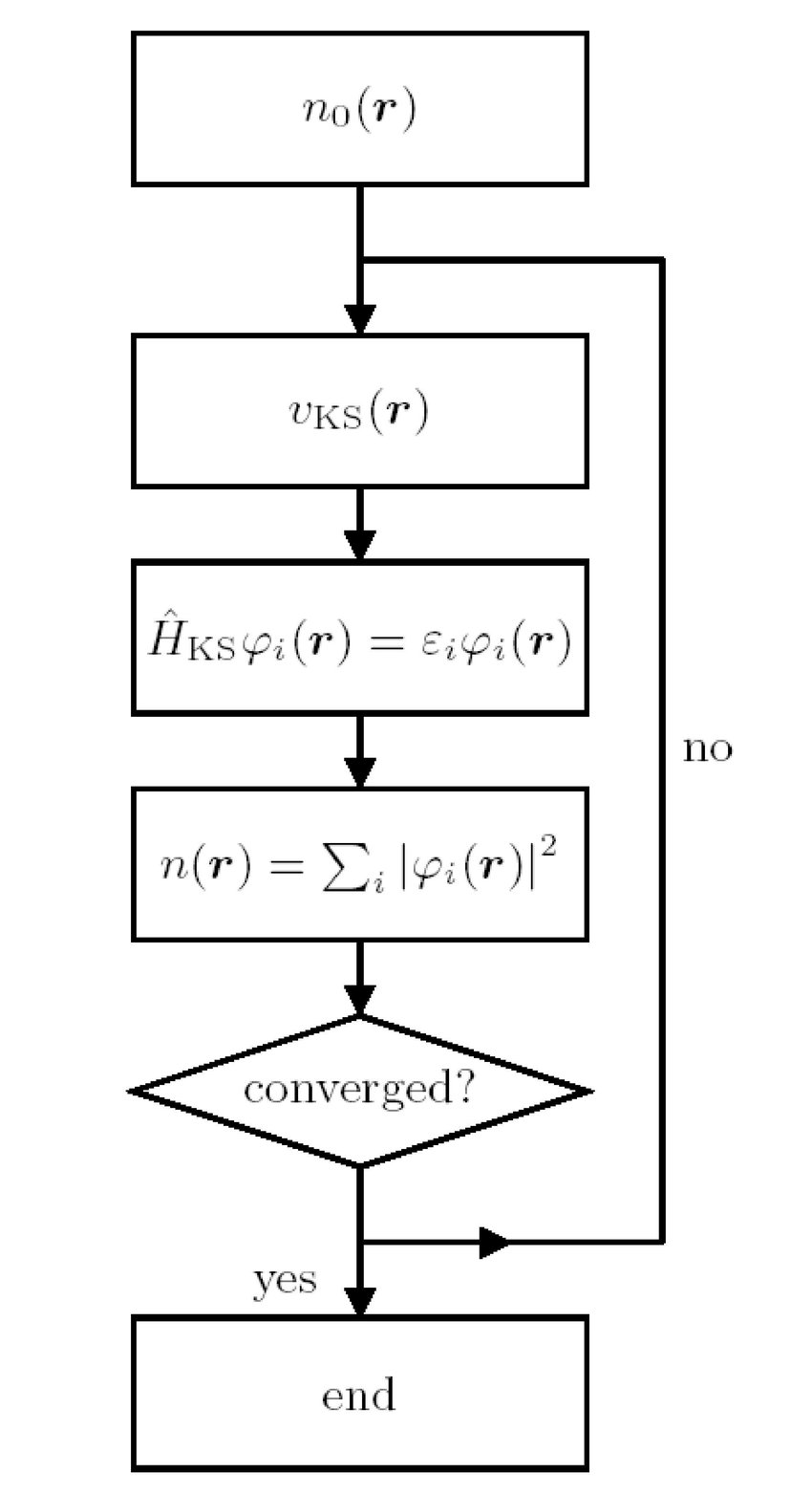
In the figure is shown the fundamental scheme of DFT self consistent algorithm. The starting point is a trial charge density that is used to calculate the Kohn-Sham potential, i.e. the potential experienced by all the other electrons in the solid. In the next step Kohn-Sham equation is solved that is nothing but a Schrodinger-like equation determined from a variational principle. If the calculated charge density is the same of the trial one then the algorithm converges and the problem is solved. Otherwise the calculated charge density becomes the input density and the cycle repeats.
In the previous weeks me and Mattia (the other guy engaged with me in the project ‘Heat transport in novel nuclear fuels‘ for IT4Innovations National Supercomputing Center at VSB – Technical University of Ostrava) worked on different materials, I studied Molybdenum, while Mattia Palladium, to understand the electronic and ionic physical property, e.g. the energy properties, the available electronic states, the structure stability, the lattice dynamics and the thermal properties. We have been working on the cluster Barbora with VASP (The Vienna Ab initio Simulation Package: atomic scale materials modelling from first principles) to run the DFT code, while we have been using Phonopy to understand the lattice vibrations (the so-called phonons).
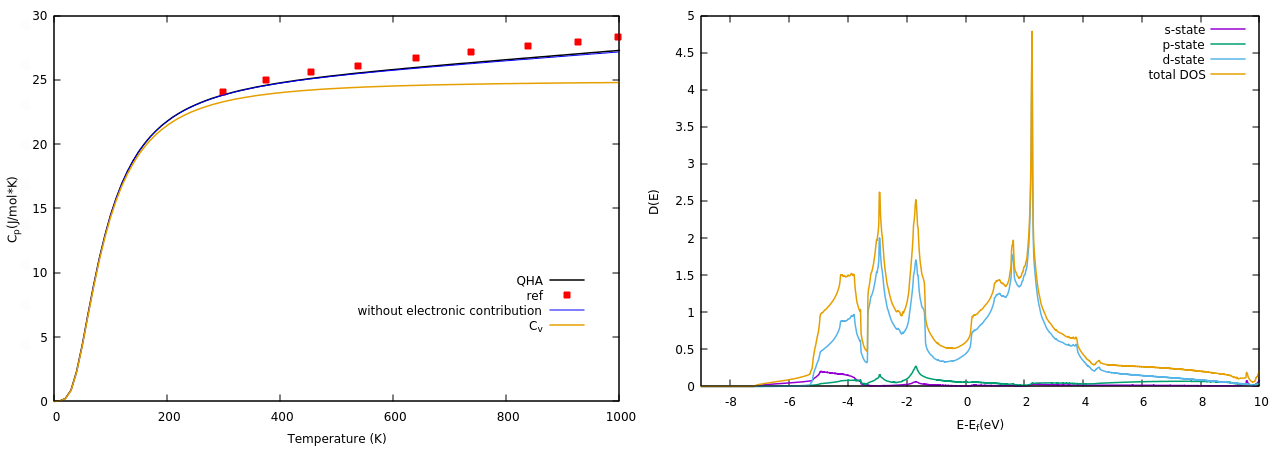
Over the next weeks we will be investigating the properties of the novel nuclear fuel. We will study the thermo-physical properties in thorium and uranium sesquicarbide (Th2C3, U2C3). In particular we will focus on how the presence of the carbon atoms affects the heat transport. If you are interested in the topic you can give a look at my next blog post, which will be focused on the procedure needed to calculate the main physical properties as thermal conductivity, heat capacity and thermal expansion that are fundamental properties in designing a new nuclear fuel. In any case, thanks for your attention and I hope you enjoyed this blog post.

Leave a Reply