Halfway Through the Journey
Hi all! In my first blog post, I briefly described the project. In this post, I will mention more about the strategy of the project. More information about project 2114 is here.
Current technology developments enable us to analyze more than 30 million genetic data points to determine their role in disease. This project will focus on re-engineering genomicper which is an R package, currently available from the CRAN repository, written before. Due to this, it can swiftly analyze larger and existing data sets.
What’s genomic permutation?
Genomic permutation, shortly Genomicper, is an approach that uses genome-wide association studies (GWAS) results to establish the significance of pathway/gene-set associations whilst accounting genomic structure. The goal of genome-wide association studies (GWAS) is to find single nucleotide polymorphisms (SNPs) that are linked to trait variation.
Project 2114 aims to re-engineer the genomicper package so it can analyse bigger and existing data sets quickly. The learning outcome of this project is to learn and implement the process of optimising a real-life piece of code.
Project Life Cycle
The general strategy cycle of the project: profile the code using data sets to determine the computational and memory expensive routines. Potentially there are 8 routines to be improved. From this profile, pick the most expensive routine to improve the efficiency, iteratively optimise the routine using microbenchmark and other optimisation techniques. More information about microbenchmark here. Test the code and compare the results produced with the original routine. Continue this cycle until code performance cannot be improved any further at which point other possible improvements will be noted, e.g. rewriting code in C, parallelisation strategies, etc. Proceed to the next routine. Once all routines have been optimised look at the strategies for further improving the code.
So Far, So Good
Thanks to the support from our mentors, we have made good progress. Until this week, the team managed to work with the lower data sets. We tried to analyze the existing performance, then optimized routines. The team tried to make the code more efficient also faster in larger data sets. While doing this, they used some of the fastest among R’s special functions. Through the analysis of the code, we attempted to make changes around the code. You can see one of our comparison results of the original code and the other version that we did in the below figure.
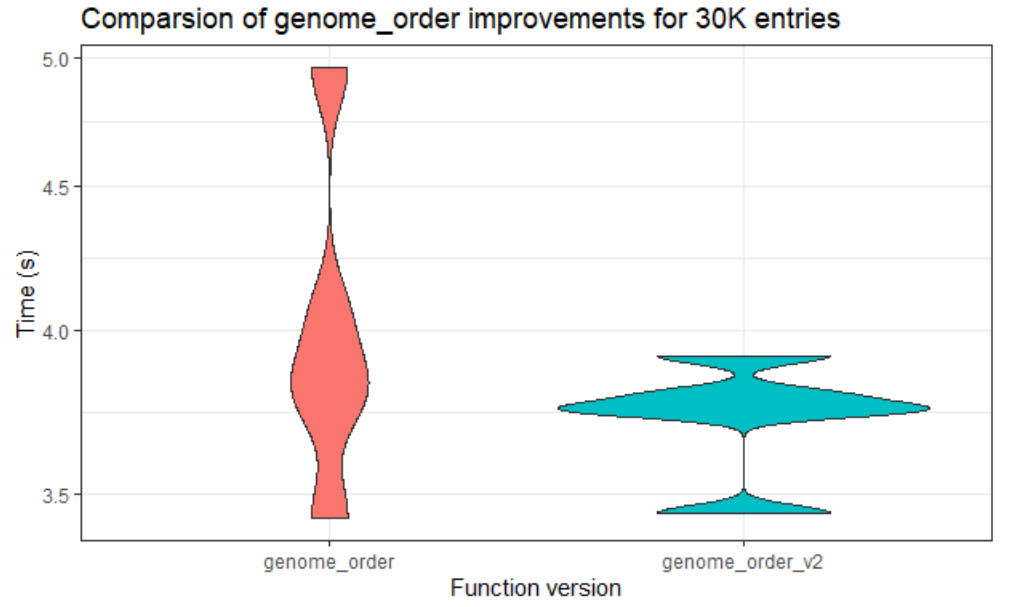
“genome_order” is the most expensive routine. We can clearly see that version 2 is faster than the original code. This helped very, especially since we made the most expensive routine efficient.
Upcoming weeks
In the upcoming weeks, all 8 routines will be optimised. After that, consider further performance improvement techniques. Once we have done all, we will try to improve the parallelisation of the code. I believe that the project process will continue well in the remaining weeks!
That is all for now. If you have any questions, I will be happy to answer.
See you on my next blog. Stay tuned!
Best,
İrem
