How to prepare MD simulation? Halfway in Athens.
First month has already passsed. That was really fast. But that is what happens when you have limited time for unlimited activities – which Greece for sure gives you, and which, for sure gives you a project in Molecular Dynamics (MD).

That is how the exemplary MD system looks like. Protein surrounded with water in a box of certain dimensions. You can see red atoms as oxygen, white – hydrogen, blue – nitrogen etc.
For those unfamiliar with the topic, MD simulation is a very powerful tool in the theoretical study of biomolecules. The simulation allows you to observe behavior of system in time, providing the description of atomic and molecular interactions which all together govern macroscopic properties of the system. That basically (and a bit oversimplifying) means that you if you have a system containing up to a few millions atoms connected with each other (e.g. protein in water) and want to see how does it change with time, you can run a simulation (usually in nanosecond scale) and can observe macroscopic changes like: conformation changes, protein interactions or folding. Visualisation
Seems easy, right? What I described above is a fun part – when the simulation is already running, HPC computers do the work for you and you don’t have to worry. There is only one condition: “garbage in, garbage out”. This is probably the most principal rule of computational chemistry – if you want a simulation to be reliable, you have to prepare it very carefully.
How do you do that? Well, as it is the first time I have dealt with this methodology and such a big system I had to learn about MD and software used for preparation and simulation first. That included reading a lot and performing a few tutorials: Schroedinger’s Maestro, NAMD, Gromacs. If you are familiar with these, you can start the real work: creating the system. What does it mean? The initial structure of the protein is obtained from structural biology studies and there are usually a few available structures for a protein. Every one of them is a bit different – they might have different ligands, different residues missing. Your aim is to search the literature and find the best one – and if none is perfect – think about how to make it perfect from homology modeling – which is basically taking what you need from different protein structures and mixing it to one.
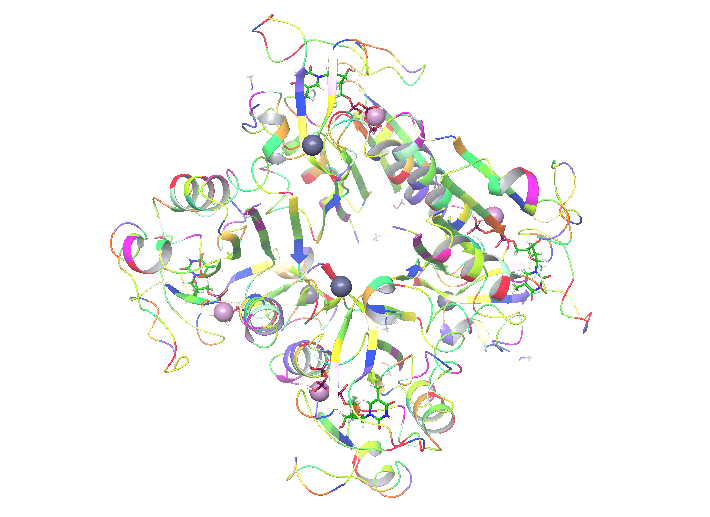
When you’re ready with the literature it’s time to put some hands on. In my project I will study: human thymidine kinase 1 enzyme (hTK1). That is one of these enzymes that none of the 3 available structures (1w4r, 1xbt and 2orv in pdb) is perfect. As I wrote above, that means that it’s my duty to make it like that with the use of proper tools. What I’ve found out is that hTK1 is the most active in its tetrameric form, and as this might be important for simulation I decided that I want to simulate it as a tetramer. Because 2orv is a dimer I rejected this structure. From the remaining two: 1w4r and 1xbt I created a homology model in Maestro. That was needed because only one of four monomers in 1w4r had a full structure of the active site and 1xbt contained essential metal atoms (Mg, Zn). Still, to make the system work we need proper ligands inside. hTK1 changes its quaternary structure upon ATP binding and that is what we want to observe, but both structures contained only the inhibiting thymidine triphosphate (TTP)
My enzyme is ready for further steps!
on the dT binding site. In order to describe both binding sites and changes upon ATP binding we had to find similar protein that would contain substrates/products or different inhibitor bounded in a way that both binding sites are solved. Here, I found similar, although not human thymidine kinase (2orw) that contained inhibitor 4TA built both from both substrate sites – deoxyadenosine and deoxythymidine, connected with 4 phosphate groups. That makes both binding sites properly occupied and the inhibitor easy to change for substrates (just delete one of the phospates) for further simulations if needed. Here, again Maestro helped to overlap the structures and change TTP to 4TA.

Visiting Aris supercomputer with Dimitris and Juan
This, along with 3 extremely intensive days in GRNET with the absolute master of MD (and patience) – Dimitris Dellis, where I’ve learnt about HPC infrastructure here and how to prepare remaining steps of simulation and run it on ARIS is what I did for last 3 weeks. Now, that the protein is ready, I have to transfer it from Maestro to Gromacs and start a simulation. That’s a plan for this week. Keep your fingers crossed! I will report how it went, soon.

Samanta, I keep my fingers crossed for you and your kinase all the time! Don’t give up and don’t forget about having a rest from time to time 😉
Thank you! We did it, and it went pretty cool. Have you seen the final presentation about the project? You can find it here: https://www.youtube.com/watch?v=5pLtKxUNQ6E
Wow, just preparing the simulation is a lot of hard work! Keep it up and I’m sure you’ll succeed:) The sky is your limit!
Ah, that’s true. Was hard. But definetely worth it! Maybe you should try applying for the program next year?