This is happening!

According to a plan, last week I’ve finally started my first proper simulation. That means, that upcoming one will finally be more chilled (hopefully). I transferred the files from Maestro to Gromacs, solvated, added ions, minimized, equilibrated (see tutorial, but don’t think that the thing is that easy for the real system. There were dozens of issues to solve on the way, of which calculating and implementing ligand charges were the least painful) and performed scaling studies to avoid waste of the CPU time. Since you can find the description of MD simulation preparation in the tutorial website, I will tell you a bit about scaling studies.
As you might remember from the last post running a simulation gives you its movement in time. That means you have some “timestep” defined along with number of steps and as an output you get your protein movement (e.g. one million of 1fs steps gives you 1ns simulation). Imagine, a typical protein with water system containing 200 000 atoms, of which every one wants to move in every of 1mln steps. That is a huge amount of data to be produced! In order to obtain any valuable results (and here value of the results depends on a time of simulation a lot – the longer simulation, the more reliable results) not only during the lifetime of a scientist, but maybe even a month or a week we have to split the whole system in smaller parts and calculate them simultaneously (parallel). This means that (warning: this sentence is a HUGE oversimplification) you can split these 200 000 atoms in smaller groups, for example 250 atoms each (100 groups) and calculate every of this group on a different core (100 cores then). Obviously, that is not really possible with the use of the desktop computer (since these usually have 4-8 cores) and this what I use the HPC infrastructure for.
Easy thing, right? Just split your system in all cores available and run it. Well. Not really a good idea. First, if you did it: you’d never get into the queue (since you’re not the only user of supercomputer these are queues to use the resources). Second: you’d get banned from the use of it, since at some point it is just the waste of time and no one (especially supercomputer admins) likes when you waste what you’re given for free (also mind the fact that you’re given a certain amount of walltime to use on a supercomputer and you sure don’t want to waste it too).
You have to remember, that splitting the system into too small parts might even slow your calculations down! That is because the parts of your system have to exchange their informations, and that also takes time – the more parts – the more time. That is mostly why you perform so called scaling studies. In my case, I decided it would be reasonable to check performance on: 100, 180, 240, 360, 480, 600, 800 and 1000 cores. I therefore performed 8 short (1ns), identical calculations on my system (differing only the number of cores used, see highlighted values in a batch script used for submitting the job) and checked how long did they take.
(…)
#SBATCH –job-name=100cores # Job name
#SBATCH –output=100cores.%j.out # Stdout (%j expands to jobId)
#SBATCH –error=100cores.%j.err # Stderr (%j expands to jobId)
#SBATCH –ntasks=100 # Number of processor cores (i.e. tasks)
#SBATCH –nodes=10 # Number of nodes requested
#SBATCH –ntasks-per-node=10 # Tasks per node
#SBATCH –cpus-per-task=1 # Threads per task
#SBATCH –time=48:00:00 # walltime
#SBATCH –mem=56G # memory per NODE
#SBATCH –partition=compute # Partition
#SBATCH –account=sohpc1601 # Accounting project(…)
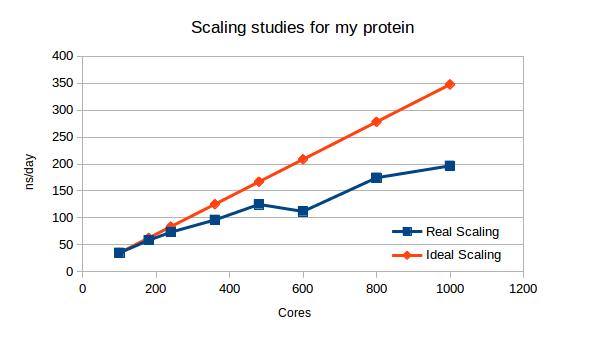
MD performance (ns/day) at different core numbers used.
Results, showed on a graph reveal that although the calculations are getting faster and faster, its acceleration is getting less and less linear and finally would reach a plateau, where the communication would take longer than calculations. At this point adding new cores is unreasonable. What you can also see there, that the biggest agreement between fast and cost-efficient solution would be running the system on 240 or 360 cores, since the deviation from ideal scaling (doubling the cores number would double the speed) is not that big there. The last conclusion: at 600 cores the system splitting is totally unfavorable, the parts might be uneven and waiting for them takes too much time.
Since I got know how to perform my calculations and the whole system is ready – I started the simulation and start a long weekend now – it’s holidays here on monday and I’m having some exciting plans for these 3 days. Next week plan: familiarizing with analytics tools available in GROMACS package, checking on a simulation, identifying and fixing problems if they occur (although I hope they won’t … pipe dream). By the end of the week I might even start comparing the system with other TK1-like enzymes. There are still lots of stones on the way and I don’t know if during the simulation I will get the conformational changes I’m hoping for, but “It’s about the journey, not the destination”. I’m learning a lot, and if my brain will not explode by the end of soHPC I will be happy.
And what do I do in order to protect my brain from exploding? Well…after (up to) 12 hours-long work day there still is a plenty of time in the evenings. And also the weekends! It’s getting harder and harder to wake up every morning, but I still manage to do that. I’ll give you some more details on life in Greece in the next post. See you soon!

{kind=link}