Woahhh we’re halfway there!

(Above is an example of a molecular dynamics simulation, where Nitrogen is the diatomic system we’re particularly interested in).
This week marks the halfway point of PRACE Summer of HPC, and we have learnt so much since starting. We have made really good progress with our project, thanks to the support from our mentors. Read below to see what we’ve been up to!
What does the project involve?
So, what does “Molecular Dynamics on Quantum Computers” actually mean? In a nutshell, we are finding the ground state energy of a diatomic system by implementing a Variational Quantum Eigensolver (VQE). This requires finding the minimum eigenvalues for the Hamiltonian operator, in the Schrodinger equation, using quantum algorithms and classical optimisation techniques to fine tune these estimates.
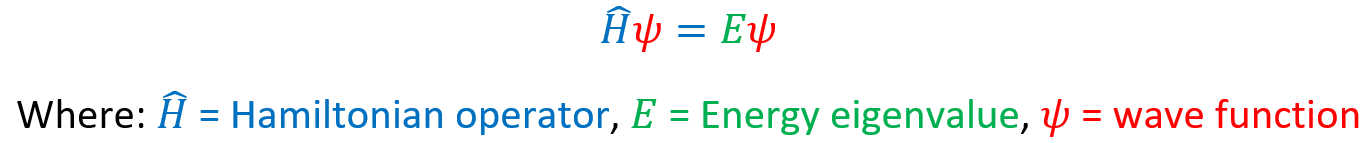
This is a highly valued application of quantum computing, as finding the ground state of molecules allows us to understand reaction rates better, and thus provides scope for drug discovery. It means that we can make predictions in quantum chemistry, without physically going into the lab and testing out different reactions. However, these simulations become increasingly more difficult as the molecules get larger and more complex. Right now, we are at the infancy stage of quantum computing – imagine the ground-breaking discoveries we can make when its full potential is unlocked!
What have we done so far?
The project is split into two main parts: VQE, and the Quasi-classical trajectories method (QCT). The aim is to join these two parts into one molecular dynamics simulation to get an accurate solution to the Hamiltonian.
The Hamiltonian acts on a wave function, and can be difficult to solve as it requires excessive computation. However, using the Born-Oppenheimer approximation, we can split the Hamiltonian into two parts (the kinetic term and the electronic Hamiltonian) and solve them separately. The separation is done based on the assumption that the nuclei is much heavier in mass than electrons, and therefore can be assumed stationary.
This week we built and finalised our VQE in Qiskit to simulate a hydrogen molecule. We initially ran the VQE on a state vector simulator, as demonstrated in IBM’s Qiskit tutorial after which, we tested the VQE on both a qasm simulator and real quantum computer. We also tested these methods using different optimisation techniques, namely COYBLA and SLSQP. The purpose of this was to compare the results against each other to find the optimum simulator and optimiser, whilst also creating artificial noise to investigate its effect on quantum estimations. One of the key challenges facing parameter optimisation is the noise associated with quantum hardware; its presence suggests that the energy calculation may not be the true objective function. Therefore, in the next few weeks, we will explore noise-mitigation techniques.
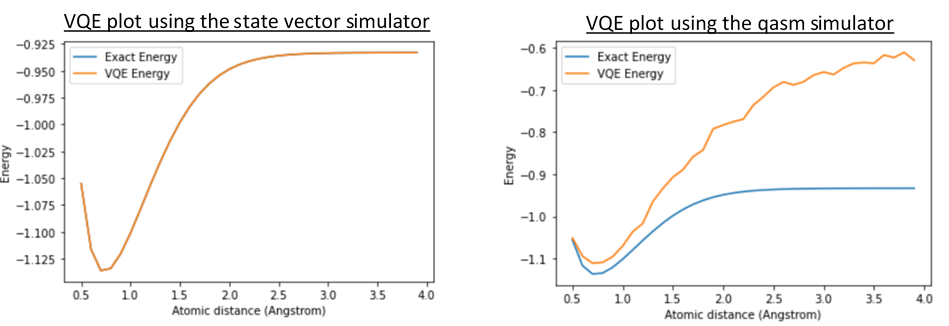
Our results above show that the state vector simulator emulates ideal execution, with the exact energy and VQE energy matching up nicely. On the other hand, the qasm simulator, which is the main Qiskit Aer backend, simulates the VQE on a real device, and includes noise models based on actual quantum hardware.
What are our targets for next week?
Next week, we will focus more on dynamics, looking at the equation of motion used for semi-classical approximations and the Born-Oppenheimer approximation, to ultimately perform calculations on the vibrations of the hydrogen molecule. However, we will first need to define a function for the numerical evaluation of the energy gradient in order to do this.
Stay tuned to hear more about our progress!
