Antiviral Drug Development with HPC
Antiviral Drug Development with MM-PBSA Calculations
This SoHPC Project aims to carry out HPC based Computational Screening for novel variants of the TAT-I24 peptide with improved binding affinity to ds-DNA.

The antiviral peptide targeted in our project is TAT-I24. The peptide TAT-I24, composed of 9-mer peptide I24 and TAT (48-60) peptide, exerts broad-spectrum antiviral activity against several DNA viruses. This peptide inhibits the replication of a large number of various double-stranded DNA viruses. Including Herpes Simplex Virus, Cytomegalovirus, Adenovirus type 5, SV40 Polyoma virus and Vaccinia virus. It aims to influence the activation of the virus by changing the amino acids of the proposed peptide and producing new variants. To further support this model, the current study explores the DNA binding properties of TAT-I24.
At the molecular level, direct binding of the peptide to viral ds-DNA can be observed. Consequently, the next simple step to further increase drug efficacy was to change any of the 22 amino acids and examine the effect of enhanced or impaired binding strength. Regarding the methods, the MM/PBSA technique can be applied. This computational approach allows semi-quantitative estimates of binding free energies (∆G) based on molecular dynamics simulations (MD).The molecular mechanics/Poisson-Boltzmann surface area (MM/PBSA) method is frequently used to calculate the binding free energy of protein-ligand complexes, regularly balancing the computational cost with attainable accuracy. The relative binding affinities obtained with the MM/PBSA approach are acceptable, but often overestimate the absolute binding free energy.
I need novel I24 variants to interact with TAT
The TAT-I24 peptide is the medicinal compound chosen in this project. It consists of the following 22 amino acid sequences, GRKKRRQRRRPPQ-CLAFYACFC. Ideally, we would test each position with the full set of 20 possible substitutions, so 2022 = 4.194304 × 1028 variants. However, individual computational connection free en-
energy calculations are absolutely inaccessible. What can be done instead is to randomly select a particular amino acid in the original sequence and replace it with another (randomly selected) and help calculate the resulting binding affinity of this newly modified peptide. MM/PBSA approach (doi:10.1021/ar000033j). In this way, at least some clues can be obtained about certain key positions in the TAT-I24 peptide that cannot be changed without compromising affinity for the target DNA. Choosing a clever strategy to substitute amino acids could reveal valuable insights into the denova design of an enhanced TAT-I24 peptide with increased activity against viral DNA.
The MM/PBSA approach has been shown to agree very well with the experimentally determined binding constants for the target system.
MD Simulations TAT-I24 in AMBER 
MD simulations and experimental protocols before MM-PBSA take time (approximately 2-3 days). For this reason, computational systems that perform as fast as possible are needed when calculating MM-PBSA.
If there is one with more speed when doing MM-PBSA :
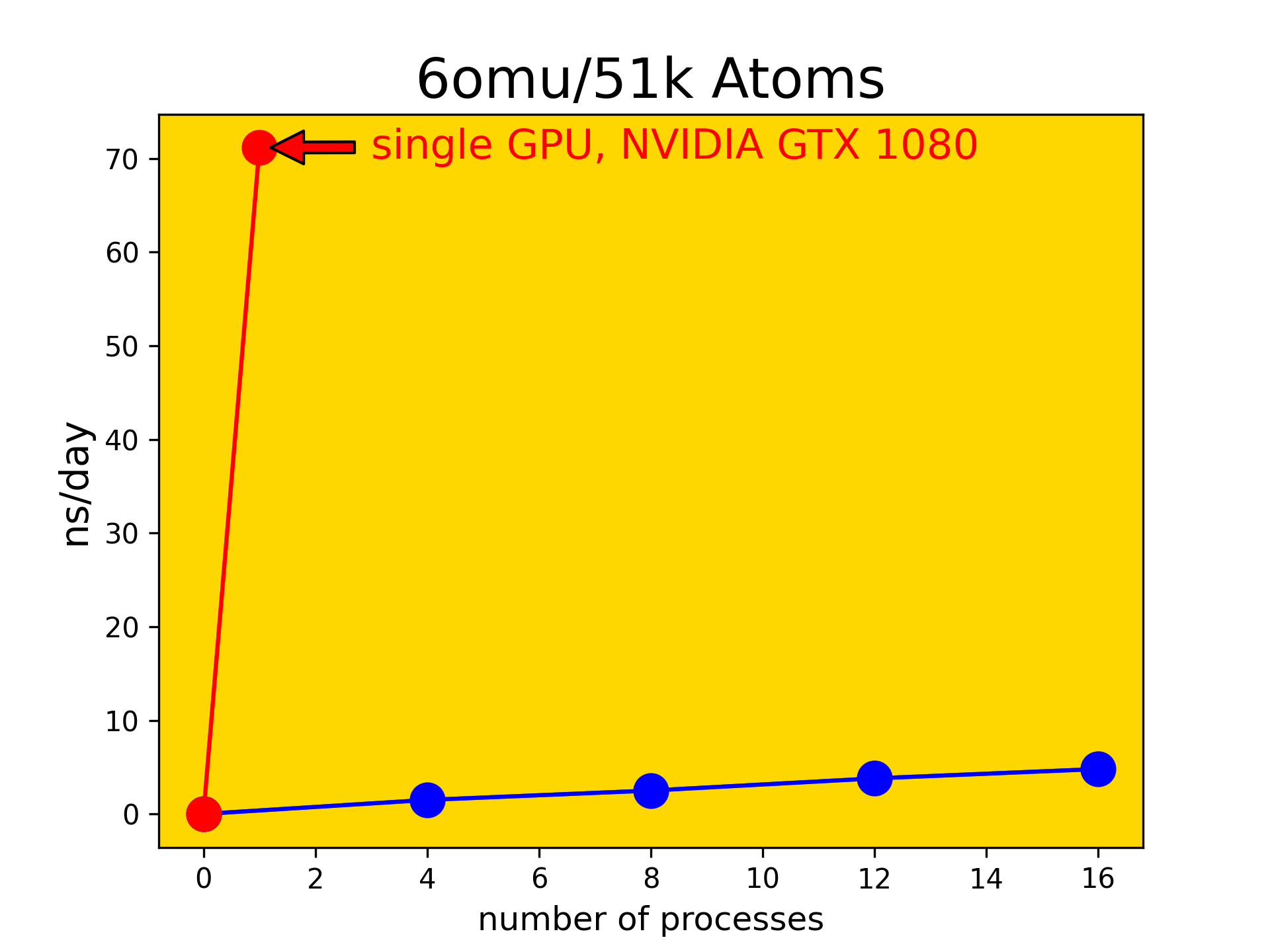
Thus we first need to establish a certain baseline of compute performance to appropriately plan and design the upcoming series of amino acid substitutions in the TAT-I24 peptide. To this end we chose an unrelated system (protein 6OMU.pdb in ex- plicit water) and carried out MD simulations with AMBER/20 using different parallel implementations. The idea was to compare conventional MPI parallel runs on CPUs to the performance available on GPUs. In this context SLURM submission scripts were set up and a series of parallel jobs was run using increasing numbers of CPUs in MPI parallel mode. The MD characteristic performance metric (ns/day) was obtained for each of these runs and plotted as function of numbers of processes (see blue dots in Fig. 2). This was then com- pared to the GPU performance avail- able on a single device of type NVIDIA geforce gtx1080 (see red dot in Fig. 2).
After comparing GPU and MPI performance, a Benchmarking was created for the experiments (with reference from previous experiments). In this way, MM-PBSA experiments are continued using GPU. In this way, more results will be obtained in a shorter time within the scope of MM-PBSA experiments using HPC.
I am doing my master's degree in Tissue Engineering and Regenerative Medicine at Bahçeşehir University / Faculty of Medicine. I also work in the Computational Biology and Molecular Simulation Laboratory (Durdagi Research Group (durdagilab.com)). Project 1: Software Based Drug Development Project: 'Discovery of Paxlovid Analogs as SARS-CoV-2 Main Protease Inhibitors'. Project 2: Structure of human Bruton's Tyrosine Kinase-associated protein virtual screening and reconstruction of target drug molecules, (Covdock-MD-MMGBSA at Schrödinger) Project 3: HPC-Derived Affinity Enhancement of Antiviral Drugs
